






نرم افزارهای مربوط به آنالیز داده های RNA-Seq جهت شناسایی توالی ترانسکریپتوم موجودات
ترانسكريپتوم مجموعه ای از RNA تولید شده (کد کننده و غیر کد کننده) در یک یا جمعیتی از سلول است که یک روش کاربردی و مهم در شناسایی ترانسکریپتوم، تکنیک RNA-Seq است.
برخی برنامه ها از قبیل Cufflinks و Scripture با اجرای محاسبات یکسان از ریدهای مستقیماً اتصال یافته به منظور شناسایی ترانسکریپتوم با روش genome- guided استفاده می کنند. نرم افزارهای Cufflinks و Scripture به ترتیب بر اساس حداکتر دقت و حساسیت عمل میکنند. روش genome- guided که در ارگانیسمهای بررسی شده در دسترس است نیازمند ژنوم مرجع نسبتاً کامل و با کیفیت بالا است.
نرم افزارهای Velvet، Trinity، rans-ABySS، و Oases (منتشر نشده) بر اساس روش – genome- independent عمل میکنند. نرم افزار Velvet دارای کارایی سرهم کردن مجدد یا دنووا ژنوم و ترانسکریپتوم است. این نرم افزار توسط یک سری الگوریتم هانقش تجزیه نمودار Bruijn و در نهایت سرهم کردن ریدها به داخل کانتیگها یا اسکافولدها را بر عهده دارد.
به طور کلی روشهای genome-guided و genome-independent به ترتیب نیازمند و فاقد نیاز به ژنوم مرجع هستند. در جدول زیر، ابزارهای لازم جهت شناسایی ترانس کریپتوم موجودات نام برده شده است.

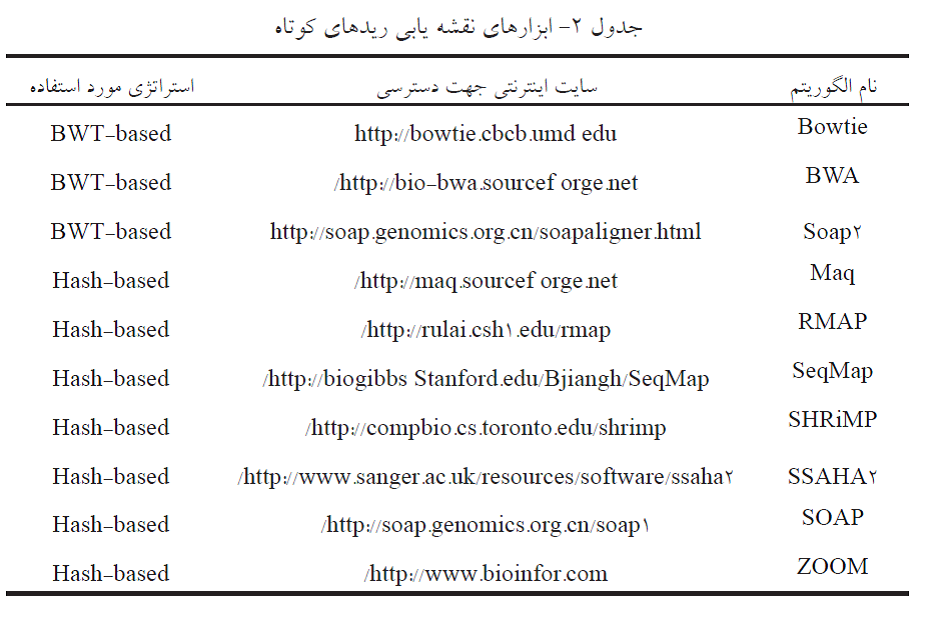
نرم افزارهای مربوط به آنالیز داده های RNA-Seq جهت نقشه یابی خوانش های کوتاه
گام پایه و بسیار مهم در آنالیز داده های RNA-Seq ردیف یابی خوانش ها است. پیچیدگی توالی های ژنوم تأثیرات مستقیم بر دقت نقشه یابی خوانشهای کوتاه دارد ، ژنـــوم پروکاریوتی به علت کوچک بودن، پیچیدگی کمتری نسبت به ژنوم یوکاریوتی دارد. ژنوم پستانداران بسیار بزرگ و محتوی توالیهای تکراری و همسان است که این مشابهت ها چالش های بزرگی برای نقشه یابی خوانش های کوتاه هستند. تفاوت زیاد در طول اگزون ها و اینترونهای یوکاریوت ها سبب دشواریهای زیادی در اجرای الگوریتم های نقشه یابی در ژنوم آنها می شود. کوتاه یا بلندی اینترون های آنها زمان محاسبات را افزایش میدهد. علاوه بر این به دلیل تعداد خوانشهای زیاد با طول 35 تا 400 جفت باز، خطاهای توالی یابی بر سختی ها و ابهامات ردیفیابی می افزاید. بر طبق نقشه یابی توالی های خوانش شده ی کوتاه، سرعت و دقت مهم ترین عامل تأثیرگذار بر داده های RNA-Seq و تکمیل نتایج تحلیلی آن به حساب میآید. برنامه های طراحی نقشه ی داده RNA-Seq با طول خوانشهای کوتاه به ۲ دسته متصل و غیر متصل تقسیم بندی می شوند. نوع متصل معمولاً برای ردیف کردن ژنوم های مرجع به خاطر وجود فواصل بزرگ ناشی از اینترونها کاربرد دارد. نوع غیر متصل مناسب برای ردیف کردن خوانش ها در مقابل مجموعه داده های رونویسی است تا بیان ژن یا ایزوفرم های ان را کمی نماید. این برنامه ها اخیراً از ۲ روش کلاسیک به طور گسترده استفاده میکنند که شامل الگوریتم های Hash و (Burrows-Wheeler Transform (BWT است. در جدول ۲، برنامه های طراحی نقشه ی داده RNA-Seq با طول خوانش های کوتاه نوشته شده است. الگوریتم های Hash-based عبارتند از Maq، ZOOM، RMAP، SeqMap و SOAP كه بر اساس كاربرد حافظه به ۲ دسته متفاوت تقسیم بندی شده است. یک نوع از این کاربردهای حافظه بر اندازه و طول ریدها و انواع دیگر بر اندازه ژنوم و طول دانه (Seed) وابسته هستند. الگوریتم های (Burrows-Wheeler Transform (BWT عبارتند از Bowtie، SOAP2 و BWA که سبب کاهش معنی دار در حافظه مورد دلخواه و افزایش معنی دار در سرعت نقشه یابی می شوند. تمامی استراتژیهای بر پایه ی BWT و Hash قادر به پردازش ریدهای کوتاه هستند ولی به خاطر روش های متفاوت در ردیف یابی خوانش های کوتاه دارای عملکرد متفاوت در کاربرد حافظه، زمان یا سرعت صرف شده، حمایت طول ریدها، تعداد خوانش های نقشه برداری شده و صحت ردیفیابی خوانش ها هستند.
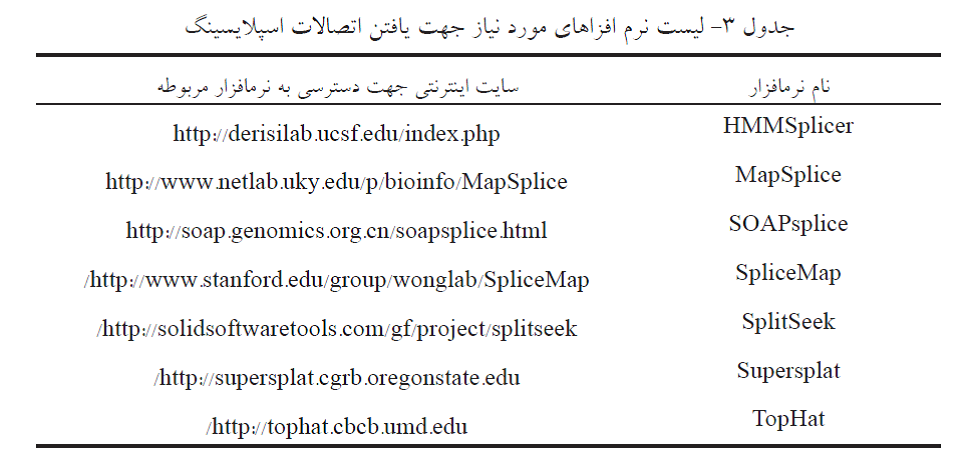
نرم افزارهای مربوط به آنالیز داده های RNA-Seq برای بررسی محل اتصال اسپلایسینگ اگزون – اگزون
اسپلایسینگ متناوب یک پدیده رایج در روند رونویسی ژنی است و اهمیت به سزایی در ساخت RNAهای ژنوم (کد کننده پروتئینی و غیر پروتئینی) دارد که کارکرد طبیعی اندام ها را نیز تضمین می کند. در حال حاضر، تعداد مدلهای موجود که به خوبی اتصال اسپلایسینگ اگزون – اگزون را تفسیر کنند بسیار کم بوده اما در جدول ۳، چندین نرم افزار که برای شناسایی محل اتصال اسپلایسینگ توسعه یافته اند نشان داده شده است.
نرم افزار TopHat در ابتدا با استفاده از الگوریتم bowtie ریدهای RNA-Seq را به ژنوم ها ردیف و سپس محل اتصالات اسپلایسینگ بین اگزون ها را بر طبق نتایج نقشه یابی پیش بینی میکند. به خاطر اینکه اکثر اینترونها دارای الگوی GT-AG هستند، نرم افزار TopHat فقط ردیف یابی خوانش های کوتاهتر از ۷۵ جفت باز را در بین تمام اینترونهای دارای الگوهای G T-AG گزارش می کند این برنامه همچنین انترونهای GC-AG و AT-AC با طول خوانش های بلندتر را جستجو خواهد نمود.
نرم افزار SpliceMap بدون توجه به تفسیر ساختارهای ژنی موجود، محل اتصالات اسپلایسینگ جدید را با دقت بالاشناسایی میکند. MapSplice نرم افزار کارآمد دیگری است که بدون وابستگی به خصوصیات محل اسپلایسینگ ور طول انترونهـــــــا میتواند محل اتصالات اسپلایسینگ را با ویژگی و حساسیت بالایی شناسایی کند. SOAPsplice نرم افزار توسعه یافته ی قدرتمندی است که قادر به شناسایی محل اتصالات اسپلایسینگ جدید بدون استفاده از اطلاعات قبلی است. این برنامه احتمالا در پیش بینی مجلد (دنوو) از محل اتصالات اسپلایسینگ و مطالعه ی اسپلایسینگ متناوب کاربرد دارد. از آنجایی که تمامی این استراتژیها نیازمند تهیه ی نقشه اولیه خوانش های RNA-Seq با ژنوم مرجع است برای موجوداتی کاربرد دارند که توالی های ژنوم مرجع آنها موجود باشد.
معرفی نرم افزارهای مربوط به آنالیز داده های RNA-Seq در سنجش بیان ژن و واریانت آن ها
روش RNA-Seq می تواند بیان ژن را در هر دو سطوح ژن و ایزوفرم های آن ارزیابی کند. در جدول 4، نرم افزارهای آنالیز بیان ژن بر اساس داده RNA-Seq گزارش شده است. نرم افزار Cufflinks در ابتدا ردیف ها را به داخل یک مجموعه رونوشت های مقرون به صرفه سر هم نموده و سپس بر اساس تعداد خوانش ها در تهیه نقشه ترانس کریپتوم، فراوانی نسبی رونوشت ها را محاسبه می کند. این برنامه همچنین می تواند ژنهای جدید و ایزوفرم های آن را بر طبق نتایج نقشه یابی خوانش ها در ژنوم مرجع پیش بینی کند. نرم افزار Scripture میتواند از ابتدا ترانس کریپتوم را نوسازی کرده و کمی کردن بیان رونوشت را انجام دهد. نرم افزار MISO (ترکیب واریانت ها) بر اساس یک چارچوب احتمالی از خوانشهای تخصیص یافته به ایزوفرم جهت ارزیابی تعداد بی-شمار ایزوفرم ها استفاده می کند. نرم افزار ALEXA-Seq ابزاری جهت آنالیز بیان متناوب و قادر به کمی کردن بیان ایزوفرم-هاست. از این رومی توان با انتخاب نرم افزارهای مرتبط آنالیز مورد نیاز بر اساس اهداف تحقیق را انجام داد. صحبت کمی نمودن بیان ژن یا ایزوفرم های آن تا حد زیادی به وسیله نتایج نقشه یابی خوانش های RNA-Seq تخمین زده شده است. ژنوم مرجع معمولاً دارای توالیهای همسان و تکراری زیادی است که در تقسیم بندی خوانش ها ابهامات زیادی ایجاد می کنند. علاوه بر این، تخصیص بندی خوانش ها در موقعیت صحیح اتصالات اسپلایسینگ بر روی ژنوم مرجع بسیار دشوار است. با توجه به این جنبه ها، دقیق ترین راه کمی نمودن بیان ژن یا ایزوفرم های آن، تهیه نقشه مستقیم خوانش های RNA-Seq از روی توالی – های محصول ترجمه (ترانسکریپتوم) است ولی بایستی به پیچیدگی ترانسکریپتوم اشاره نمود که ساختن پایگاه داده دقیق و کامل از آن حتی در گونه هایی که مطالعات خوبی روی آنها انجام شده (انسان و موش) کاری بس دشوار است

جدول 4.
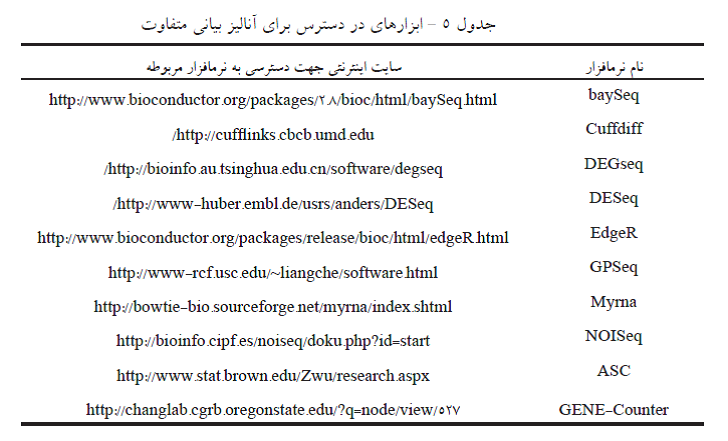
معرفی نرم افزارهای مربوط به آنالیز داده های RNA-Seq برای یافتن واریانت های مختلف ژن ها
ژن های یوکاریوتی تحت شرایط مختلف و تامین نیاز موجودات، چندین ایزوفرم متمایز را نشان خواهند داد. با تعیین تغییرات بیان ژنها یا ایزوفرم های آن بین ۲ نمونه متفاوت، انجام آنالیزهای بیانی متفاوت جهت شناسایی ژنها یا ایزوفرم های بیان شده امکان پذیر خواهد شد. در جدول ۵، یکسری استراتژی ها تحت نرم افزارهای مختلف موجود است که قادرند با استفاده از دادههای RNA-Seq ژنها یا ایزوفرم های بیان شده را شناسایی کنند. این روشها به ۲ دسته مدل های پارامتریک و غیر پارامتریک تقسیم بندی می شوند. روشهای پارامتریک بر اساس توزیع های احتمالی شناخته شده (دوجمله ای، پواسون و دوجمله ای منفی) و غیر پارامتریک فاقد فرضیات توزیع داده هستند. تارازونا و همکاران (۲۰۱۱)، مدل NOISeq (روش غیر پارامتریک قدرتمند) را برای برطرف کردن اریب داده های RNA-Seq از تغییرات عمق توالی یابی پیشنهاد کردند ، نتایج آزمایشات آنها نشان داد که این مدل نسبت به روش های پارامتریک DESeq ، baySeq و edger که برای توزیع دوجمله ای منفی استفاده می شوند، در مقابل تغییرات عمق توالی یابی انعطاف پذیرتر است در ضمن وی اثبات کرد که روش های پارامتریک موجود نسبت به NOISeq به عمق توالی یابی بالا وابسته هستند.