









دانلود فایل آموزش نرم افزار مگا ( mega 6) جهت ترسیم درخت فیلوژنتیکی به فرمت PDF در پایین صفحه
آموزش نرم افزار مگا (mega) جهت ترسیم درخت فیلوژنتیکی
نرم افزار MEGA یکی از نرم افزار های فوق العاده و پرکاربرد بیوانفورماتیک که جهت آنالیز فیلوژنتیکی توالی های DNA و پروتئینی و ترسیم درخت های فیلوژنتیکی مورد استفاده قرار می گیرد. جهت دانلود نرم افزار مگا کلیک نمایید.
کاربرد اول نرم افزار mega: در مثال اول، یکی از روش های دسته بندی موجودات و ترسیم درخت فیلوژنتیکی استفاده از توالی های DNA و پروتئین های همولوگ می باشد. روش کار به این صورت است که ابتدا از چند فرد، گونه ویا جمعیت نمونه گیری صورت میگیرد که نیاز به خوشه بندی این افراد است. قدم بعدی تعیین ژن های همولوگ به عنوان یک مارکر ژنتیکی برای خوشه بندی جمعیت می باشد. که این قطعه تکثیر شده و سپس توالی یابی می گردد. حال میتوان از این توالی ها برای خوشه بندی افراد و ترسیم درخت فیلوژنتیکی استفاده نمود.
کاربرد دوم: در تحقیقات خود یک موجود یا گونه و یا جمعیت جدید را مورد بررسی قرار میدهید و یا جدیدا کشف نموده اید و میخواهید گروه و یا زیرگروه (clade) این موجود را تعیین کنید. مانند روش فوق یک ژن همولوگ انتخاب نموده و بعد از تکثیر آن و توالی یابی، توالی ژنوم این موجود جدید را توالی های موجودات شناخته شده در پایگاه های داده مقایسه ودرخت فیلوزنتیکی آن را ترسیم میکنید تا تعیین شود این موجود در کدام خوشه یا دسته قرار میگیرد.
کاربرد سوم: چند ژن با توالی مشابه در انسان را در نظر بگیرید (مانند خانواده ژنی هموگلوبین ها). حال میخواهید سیر تکاملی این ژن ها را بررسی نمایید بدین صورت که کدام ژن قدیمی تر و به نوعی منشا سایر ژن ها می باشد؟ کدادم ژن از کدام ژن مشتق شده است؟ کدام ژن جدیدتر است؟ با ترسیم درخت فیلوزنتیکی میتوان ان را بررسی نمود
کاربرد چهارم: با استفاده از توالی های همولوگ میخواهید که تکامل گونه ها را بررسی نمایید.
آشنایی با اجزای درخت فیلوژنتیکی

Branch: یا شاخته همان افرادی هستند که در ترسیم درخچه وجود دارند و درخت را برای دسته بندی ان ها ترسیم میکنیم. چند شاخه تشکیل یه خوشه یا دسته را میدهند.
Node: محل اتصال شاخه ها ویا خوشه ها به یکدیگر را گویند
Ingroup: فرض کنید میخواهیم در یک خانواده چند ژن تعیین کنیم کدام ژن قدیمی تر و کدام جدیدتر است. برای این کار در ترسیم درخت از یک ژن دیگر خارج از خانواده ژنی مورد نظر استفاده میکنیم بنابراین بعد از ترسیم درخچه هر ژنی که به آن ژن خارجی نزدیک بود دارای قدمت بیشتری می باشد. به خانواده ژنی Ingroup و به ژن خارج از خانواده Outgroup گویند.
Outgroup
Root: یا ریشه، به محل اتصال Outgroup به Ingroup ریشه گفته میشود چون سبب میشود سایر ژن ها نسبت به Outgroup سازماندهی کنیم.
آموزش کاربردی نرم افزار مگا
درس اول: تعیین ساختار تکاملی هموگلوبین ها در انسان
آموزش نحوه وارد نمودن توالی ها به نرم افزار مگا 6 و 7
نحوه یافتم توالی ژن ها در ncbi
در انسان خانواده هموگلوبین ها شامل , HBA1, HBA2, HBB, HBD, HBE1, HBG1, HBG2, HBM, HBQ1, HBZ می باشد که میخواهیم از درخت فیلوژنتیکی این زن ها را برای بررسی تکامل آن ها بررسی نماییم. در این کار ما به یک Outgroup نیاز داریم که از میوگلوبین( , MB) استفاده میکنیم.

شکل 2: قدم اول: در یافت توالی پروتئینی این ژن ها از پایگاه داده NCBI. برای این کار به آدرس https://www.ncbi.nlm.nih.gov/gene/?term=HBA1 رفته و ژن ها را یک به یک جستجو می نماییم. و سپس روی ژن انسانی کلیک میکنیم ( در این فهرست همین ژن مربوط به سایر موجودات نیز لیست شده اند)

شکل 3: در پایین صفحه باز شده سمت راست عبارت RefSeq Proteins را پیدا نموده و روی آن کلیک کنید تا توالی پروتئین این ژن نمایش داده شود.

شکل4: در این صفحه ساختار و ویژگی های پروتئین مورد نظر نشان داده شده است . در صفحه باز شده برروی عبارت FASTA کلیک کنید تا توالی پروتیئن نشان داده شود.

شکل 5: توالی پروتئین مورد نظر به فرمت FASTA
فرمت FASTA به حالت زیر است. در ابتدا حتما باید علامت > را داشته باشد بلافاصله بعد این علامت اسم پروتیئن و ژن می آید که این مشخصات میتواند در چندین خط باشد اما حتما باید در یک پاراگراف باشند (قسمت آبی رنگ). و سپس در پاراگراف بعدی( با زدن کلید ENTER در صفحه Word) توالی پروتیئن ذکر می شود. بنابر این با رعایت سه اصل فوق می توانید برای هر توالی ای یک فرمت FASTA بسازید که در ترسیم درخت فیلوژنتیکی بسیار کاربرد دارد.


شکل 6: توالی ژن اول را کپی کرده و در یک notepad دخیره نمایید (با فرمت FASTA) سپس به سراغ زن بعدی بروید و توالی تمام ژن ها را در همان فایل notepad و در زیر یکدیگر دخیره نمایید( بین توالی هر ژن با زدن دکمه ENTER کیبورد چند فاصله ایجاد کنید. تعداد فاصله ها مهم نیست)

شکل 7: نحوه وارد کردن توالی ها در یک فایل. در صورتی که چند توالی دارید که میخواهید آن ها را بررسی نمایید همه توالی ها را به فرمت .fasta در آورید و مانند شکل فوق در یک فایل notepad قرار دهید.


شکل 8: سپس فایل را به صورت بالا ذخیره کنید. در قسمت save as a type گزینه All file (*.*) را انتخاب نمایید و در قسمت File name اسم فایل را seq.fasta بگذارید تا فایل فوق با فرمت .fasta ذخیره شود.
در صورتی که بخواهید ژنی را به بررسی اضافه و یا حذف نمایید از ظریق لیست فوق اقدام به ویرایش تعداد ژن ها نمایید.
آموزش وارد کردن توالی ها در نرم افزار mega
در صورتی که نرم افزار مگا را نصب کرده باشید و فایل توالی ها را با فرمت .fasta دخیره کرده باشید با باز کردن فایل ( دبل کلیک روی فایل) مستقیم توالی ها وارد نرم افزار مگا می شوند. در غیر این صورت به این گونه عمل کنید:

نرم افزار مگا را باز نمایید و مراحل را به ترتیب مانند شکل های زیر دنبال نمایید.


چون توالی ما پروتئین است در این قسمت گزینه پروتئین را انتخاب میکنیم

حال در پنجره Alignment Explorer (شکل فوق) به طریقه زیر فایل حاوی توالی خود را وارد می کنیم


سپس به فولدر حاوی فایل بروید و فایل را انتخاب و سپس open نمایید.

آموزش هم ردیف نمودن (alignment) توالی ها در نرم افزار مگا 6 و 7 (mega 6, 7)
اگر مراحل فوق را به درستی طی کرد باشید توالی های پروتئین ها در صفحه Alignment Explorer بار گذاری می گردد (شکل فوق). قبل از ترسیم درخت فیلوژنتیکی باید تمام توالی ها هم ردیف شوند. برای این کار به روش زیر اقدام فرمایید.
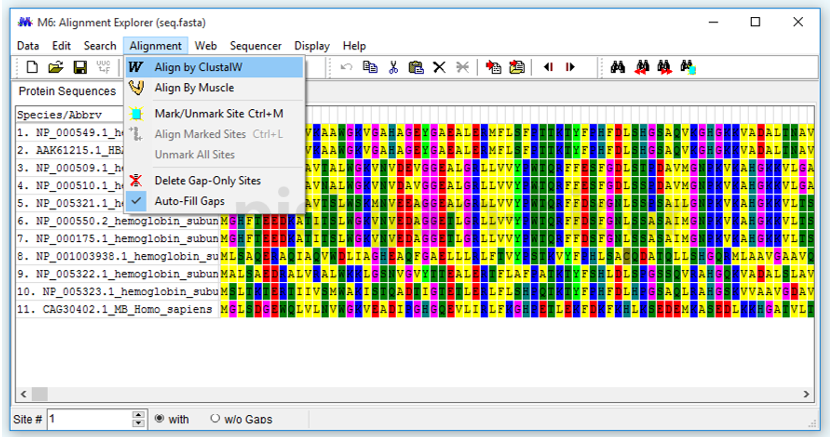



نرم افزار در حال هم ردیف کردن توالی ها (شکل فوق)
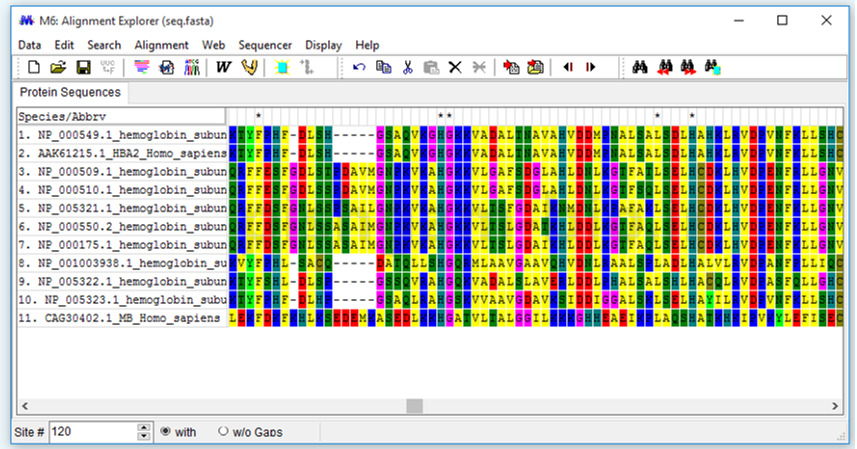
حالت توالی ها بعد از هم ردیف شدن توالی ها. در این حالت شکاف هایی بین توالی های پروتئین ها مشاهده میگردد که کاملا طبیعی می باشد. برای ترسیم درخت فیلوژنتیکی مراحل بعدی را دنبال نمایید.
آموزش ترسیم درخت فیلوژنتیکی بوسیله نرم افزار مگا 6 و 7


سپس مانند شکل فوق به صفحه اصلی نرم افزار برگردید ( صفحه قبلی را نبندید). در این حالت میتوانید اقدام به ترسیم درخت فیلوژنتیکی نمایید.

از منوی phylogeny نوع درختی را که میخواهید ترسیم کنید انتخاب نمایید. در این شکل درخت Neighbor-Joining انتخاب شده است.

در این شکل گزینه yes را انتخاب نمایید.

در شکل فوق باید پارامتر هایی همچون ضریب تشابه بین افراد (گزینه mode/method) و نوع آزمون درستی درختچه (گزینه test of phylogeny) را انتخاب نمایید. در این حالت میتوانید گزینه های دیفالت خود نرم افزار یا قبول نمایید یا براساس مقالات پارامترهای خود را وارد نمایید. سپس در نهایت گزینه compute را کلیک نمایید تا درخت شروع به ترسیم شود.

نرم افزار در حال انجام آنالیزهای ترسیم درخت

شکل فوق: درخت رسم شده. همان گونه که مشاهده می فرمایید ژن MB که به عنوان outgroup استفاده شده است در یک شاحه مجزا نسبت به بقیه قرار گرفته است و درخت را ریشه دار (rooted) نموده است و درخت دارای دو خوشه مجزا می باشد. اعدادی که در node ها نوشته شده اند مربوط به آزمون درستی ترسیم درخت می باشد که هر چه به 100 نزدیکتر باشد خوشه بندی قویتر خواهد بود. برای دریافت اطلاعات مربوط به درخت رسم شده روی منوی caption کلیک نمایید.

شکل فوق: اصلاعات مربوط به ترسیم درخت که میتوان به عنوان توضیح در زیر درخت آن ها را افزود.
آموزش محاسبه ضریب فاصله ژنتیکی بین نمونه ها و جمعیت ها بوسیله نرم افزار مگا
برای محاسبه ضریب فاصله ژنتیکی بین افراد مورد بررسی از روش زیر استفاده میشود



تعیین نوع ضریب درخواستی و سپس کلیک برروی compute.

ضرایب فاصله بین افراد مورد بررسی.
آموزش تعیین کلاد نمونه ها بوسیله نرم افزار مگا
درس دوم: تعیین گروه یا زیرگروه (clade) ژنتیکی افراد مورد بررسی
فرض کنید در تحقیقات خود یک موجود یا گونه و یا جمعیت جدید را مورد بررسی قرار میدهید و یا جدیدا کشف نموده اید و میخواهید گروه و یا زیرگروه (clade) این موجود را تعیین کنید. برای ان کار معمولا از توالی هایی مانند its, cp23s و سایر RNAهای ریبوزومی استفاده میشود. بعد از تکثیر ژن مورد و توالی یابی آن، ما دارای چند فایل توالی هستیم که باید توالی ژنوم این موجود جدید را توالی های موجودات شناخته شده در پایگاه های داده مقایسه نموده ودرخت فیلوزنتیکی آن را ترسیم نماییم تا تعیین شود این موجود در کدام خوشه یا دسته قرار میگیرد. برای مثل برای یک گونه 8 گروه ژنتیکی از A تا H وجود دارد حال افراد مورد بررسی ما به کدامین گروه ژنتیکی تعلق دارند؟
در مرحله اول باید توالی موجود در فایل های تعیین توالی را مورد خوانش قرار دهیم. باز نمودن فایل های توالی با نرم افزار مگا امکان پذیر می باشد اما یک نرم افزار مناسب برای این کار نرم افزار chromas میباشد.
آموزش نرم افزار کروماس (chromas)
چگونگی کار با فایل های تعیین توالی (آشنایی با نرم افزار chromas).

شکل 1: فرض کنیم که شما قطعه خود که از طریق PCR تکثیر شده است را برای تعیین توالی فرستاده اید و فایل تعیین توالی را دریافت نموده اید. فایل های تعیین توالی دارای پسوند .abi در اخر خود هستند. حال باید با استفاده از این فایل توالی خود را استخراج نماییم. برای این کار باید از نرم افزار Chromas استفاده نمود

شکل 2: نمای کلی نرم افزار کروماس. برای وارد کردن توالی در آن باید از گزینه open استفاده نمود و سپس از پنجره ای که باز میشود فایل توالی خود را وارد نماییم.

شکل 3: شکل توالی باز شده توسط نرم افزار کروماس. در بالا توالی قطعه ژنوم و در پایین پیک های هر نوکلئوتید را مشاهده میکنید. توجه کنید که 40 نوکلنئوتید ابتدایی هر توالی معمولا داری پیک های نا مشخص می باشند بنابراین نباید به توالی 40 نوکلئوتید ابتدایی توجه کرد. همین طور در قطعات بزرگ نیز معمولا از نوکلئوتید حدود 600 به بعد پیک ها حالتی نا مشخص میگیرند به خود میگیرند. پس در گام اول توالی را با نرم افزار کروماس بررسی میکنیم تا دارای پیک های واضح و جداگانه باشد در غیر این صورت نمی توان به توالی اعتماد کرد.

شکل 4: نمونه ای از پیک های به هم ریخته و غیرقابل اعتماد

شکل 5: نمونه ای از پیک های به هم ریخته و غیرقابل اعتماد در ابتدا (شکا بالا 25 نوکلئوتید اول) و انتهای (شکل پایین) یک فایل توالی یابی

شکل 6: برای استخراج توالی از گزینه file گزینه bach export را انتخاب میکنیم تا صفحه زیر باز شود.

شکل 7: در پنجره باز شده فایل توالی را انتخاب میکنیم و سپس بدون تغییر سایر گزینه ها روی Export کلیک میکنیم. در همان فولدری که فایل توالی یابی قرار دارد توالی استخراج گردد.

شکل 8: توالی استخراج شده را می توان برای ترسیم درخت فیلوژنتیکی به کار برد. اما ابتدا باید نوکلئوتید های مشکوک اول و اخر توالی حذف گردند و سپس فایل به فرمت FASTA در آید.
حال که ما توالی های مربوط به افراد جمعیت خود (با گروه بندی ژنتیکی نامشخص) را داریم باید توالی های مربوط به افرادی که گروه ژنتیکی ان ها مشخص است نیز از پایگاه های داده دریافت گردد. بهترین گزینه برای دریافت این اطلاعات استفاده از نرم افزار Blast در پایگاه داده NCBI می باشد.
نحوه یافتن باید توالی های مربوط به افراد با گروه ژنتیکی مشخص
برای انجام این بلاست به صفحه:

شکل فوق: صفحه اولیه انجام nucleotide blast به این شکل می باشد. که بررسی این صفحه در شکل های بعدی خواهیم پرداخت.

شکل فوق: در این کادر توالی حاصل از تعیین توالی افراد خود را وارد می نماییم

شکل فوق: در این کادر تعیین میکنیم که جستجو در چه نوع توالی هایی انجام بپذریرد که ما گزینه زیر را انتخاب میکنیم : Nucleotide collection (nr) که شامل تمام توالی هایی که در پایگاه داده NCBI ثبت شده است مانند توالی ژنومی و mRNA ها و سایر RNAها می گردد

شکل فوق: در این قسمت باید نام گونه ای که میخواهیم در آن جستجو کنیم وارد کنیم. در صورتی که هیچ گونه ای وارد نشود جستجو در تمامی موجودات انجام می پذیرد.

شکل 18: در ناحیه باید میزان شباهت بین دو گونه یکی از این سه گزینه را انتخاب نمایید.
Highly similar sequences (megablast): در صورتی که جستجو در یک گونه انجام می پذیرد. چون توالی ما و توالی های یافت شده بسیار به هم شبیه هستند(بیشتر از 75 درصد شباهت بین دو توالی) از این گزینه استفاده میکنیم.
More dissimilar sequences (discontiguous megablast): در مقایسه دو گونه نزدیک به هم استفاده می شود یا حالتی که توالی ما و توالی های یافت شده مقداری از هم تفاوت دارند (بین 60 تا 75 درصد شباهت بین دو توالی).
Optimize for Somewhat similar sequences (blastn): برای مقایسه بین گونه های دور از هم و یا حالتی که توالی ما و توالی های یافت شده از هم تفاوت (بیش از 50 درصد تفاوت) دارند.
بعد از انجام تنظیمات فوق برروی گزینه BLASTکلیک می کنیم.

شکل فوق: هنگامی که عملیات بلاست در کامپیوتر های پایگاه داده NCBI در حال انجام است این صفحه به نمایش در می اید که به صورت اتوماتیک نوسازی می گردد. تا انجام کامل عملیات این صفحه به صورت خودکار به صفحه نتایج منتقل میگردد.

شکل فوق: در صفحه نتایج ، این نمایشی گرافيکی از نتایج BLAST است به طوری که ١٠٠ توالی اولی که در جستجوی BLAST به دست آمده اند به صورت خطوط رنگی نشان داده می شوند هر توالی بر اساس ميزان شباهت خود دارای یک طيف رنگی است .کليد رمز رنگ ها بر اساس امتياز همرديفي در بالای آن آمده است. هر چه قدر جور شدن توالي يافت شده با توالي در حال جستجو بيشتر باشد با رنگ قرمز و هر چه کمتر باشد با رنگهای رو به مشکی نمایش داده می شود. بنابراین در این قسمت در یک نگاه کلی می توانيم ميزان شباهت را مشاهده کنيم .

شکل فوق(این شکل جهت یادگیری نرم افزار blast می باشد و در ترسیم درخت نقشی ندارد): در بخش سوم شماره دسترسي (شماره 7) و نام( شماره 1) توالی های بدست آمده فهرست شد هاند. روبروي هر توالی دو عدد هست. اولين عدد امتياز همرديفي دوگانه (شماره 3 و2) توالي يافت شده و توالي در حال جستجو است که آليه اطلاعات بعدي بر اساس آنها مرتب می شوند. دومين شاخص، ارزش مورد E-value (شماره 5) می باشد که برای قابل قبول بودن توالی یافت شده باید این عدد کمتر از 0.01 باشد، در غیر این صورت جفت شدن توالی ها تصادفی می باشد. شماره 6 میزان شباهت دو توالی را شان میدهد، شماره 4 درصدی از توالی ها که با هم همپوشان هستند را نشان میدهد.

با توجه به معیارهای ذکر شده مانند E-value و میزان همپوشانی (بالای 50درصد) ما افرادی را که گروه گروه زنتیکی آن ها مشخص است انتخاب مینماییم و مربع کنار آن ها را تیک میزنیم مانند کادر قرمز رنگ. بعد از این کار ما باید توالی این این افراد را دانلود نماییم برای این کار مانند شکل زیر عمل نمایید.

بعد از این مرحله یک فایل با فرمت .fasta دانلود میشود که حاوی اطلاعات مربوط به افراد با گروه های ژنتیکی شناخته شده می باشند. این فایل را با فایل حاوی توالی های افراد مورد بررسی خود ادغام مینماییم و به عنوان یک فایل واحد جهت ترسیم درخت فیلوزنتیکی مورد استفاده قرار میدهیم. حال هر فرد مورد بررسی ما در هر خوشه ای کنار افراد با گروه زنتیکی خاص قرار بگیرد، این فرد نیز دارای همان نوع گروه زنتیکی می باشد.
در صورتی که میخواهی از توالی پروتئین ها برای ترسیم درخت فیلوزنتیکی استفاده کنیم برای بلاست توالی خود از این صفحه نرم افزار بلاست که مخصوص پروتئین ها می باشد استفاده میکنیم: https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastp&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome
آموزش باز نمودن فایل های abi با نرم افزار مگا
درس سوم: وارد نمودن مستقیم داده ها از فایل های توالی یابی به نرم افزار mega
برای وارد نمودن داده های توالی یابی به نرم افزار مگا از روش زیر استفاده نمایید:



برای افزودن توالی فوق به پنجره Alignment Explorer مانند زیر عمل نمایید

با استفاده از روش فوق میتوانید همه داده های موجود در فایل ها توالی را به پنجره Alignment Explorer وارد نموده و سپس اقدام به ترسیم درخت فیلوژنتیکی نمایید.

نحوه ذخیره داده های موجود در فایل توالی یابی به فرمت .fasta
[divider]
دانلود فایل آموزش نرم افزار مگا ( mega 6) جهت ترسیم درخت فیلوژنتیکی
دیدگاهتان را بنویسید