










معرفی نرم افزار پاپ ژن (popgene)
popgene نرم افزار محبوب و بسیار مفید برای آنالیز داده های ژنتیک جمعیت است. این نرم افزار تنوع ژنتیکی بین و درون جمعیت ها را با استفاده از مارکرهای همبارز و غالب، داده های دیپلوئید و هاپلوئید و همچنین داده های صفات کمی آنالیز می کند. این نرم افزار آماره هایی مانند فراوانی آللی، تنوع ژنی، فاصله ژنتیکی، آماره های F، ساختار چندلوکوسی و …را محاسبه می کند.
POPGENE is a user-friendly Microsoft® Window-based computer package for the analysis of genetic variation among and within natural populations using co-dominant and dominant markers and quantitative traits. This package provides the Windows graphical user interface that makes population genetics analysis more accessible for the casual computer user and more convenient for the experienced computer user. Simple menus and dialog box selections enable you to perform complex analysis and produce scientifically sound statistics, thereby assisting you to adequately analyze population genetic structure using the target markers/traits.
These software is designed specifically for the analysis of co-dominant and dominant markers using haploid and diploid data. It performs most types of data analysis encountered in population genetics and related fields. It can be used to compute summary statistics (e.g., allele frequency, gene diversity, genetic distance, F-statistics, multi locus structure, etc.) for (1) single-locus, single populations; (2) single-locus, multiple populations; (3) multi locus, single populations and (4) multi locus, multiple populations.
آموزش مارکر های زیستی هم بارز (Co-Dominant)، غالب (Dominant) و کمی (Quantitative) چیست
انواع مارکر های زیستی که توسط این نرم افزار میتوان بررسی نمود:
Co-Dominant: در این حالت می توان افراد هتروزیگوت را از هموزیگوت ها تشخیص داد مانند مارکر های SSR، AFLP و RFLP
Dominant: در این حالت نمیتوان فراد هتروزیگوت را از هموزیگوت ها تشخیص داد مانند مارکر های ISSR، RAPD
Quantitative: صفات کمی مانند اندازه محتوای DNA افراد
انواع ژنوتیپ های مورد بررسی در این نرم افزار
هاپلوئید: برای موجودات هاپلوئید
دیپلوئید: برای موجودات دیپلوئید (و سایر موجودات غیر دیپلوئید)
آموزش نحوه آماده سازی داده های مارکرهای Co-Dominant مانند SSR، AFLP و RFLP برای نرم افزار popgene
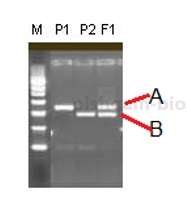
در این شکل ما یک مارکر SSR را بررسی نموده ایم که داری دو آلل است و فرد مورد بررسی نیز دیپلوتایپ است. در این بررسی سه فرد وجود دارد که عبارتند از P1, P2 و F1 . حال میخواهیم داده های این سه نفر را در جدول وارد نماییم. آلل بالا را A والل پایین را B می نامیم پس ژنوتیپ افراد به این صورت میشود:

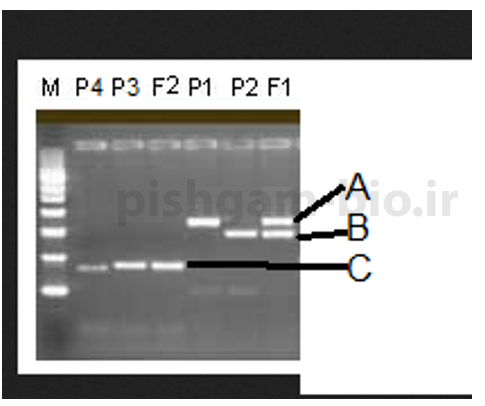
در این شکل ما یک مارکر SSR را بررسی نموده ایم که داری 3 آلل است و افراد مورد بررسی نیز دیپلوتایپ هستند، پس هر فرد فقط دو الل دارد. در این بررسی 6 فرد وجود پس ژنوتیپ افراد به این صورت می شود:

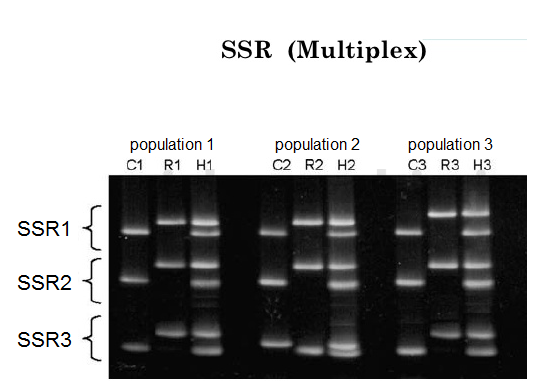
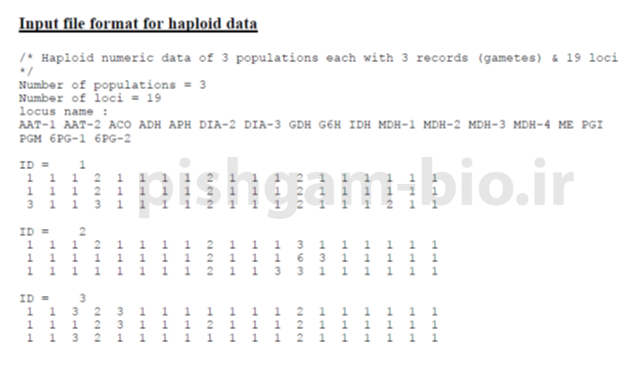
در این شکل ما با سه مارکر SSR( شامل لوکوس های SSR1, SSR2, SSR3) سه جمعیت را مورد بررسی قرار داده ایم که هر جمعیت شامل 3 فرد است. پس در مجموع 9 فرد در سه جمعیت داریم. برای شکل فوق میخواهیم یه فایل ورودی داده برای نرم افزار popgene درست نماییم.
در ابتدا مشخص نماییم هر الل را چه بنامیم. می توان در هر لوکوس به صورت توافقی الل بالا را A و الل پایین را B بنامیم و برای هر سه لوکوس از علامت های A وB استفاده نماییم. برای نرم افزار فرقی نمیکند چون هر لوکوس را جداگانه مورد بررسی قرار میدهد. پس ژنوتیپ افراد به این صورت می شود.

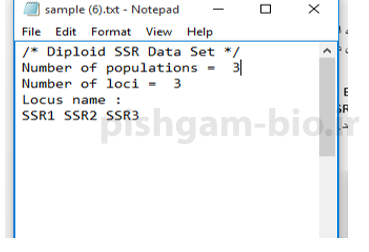
شکل فوق: برای فایل ورودی داده برای نرم افزار popgene یک فایل Notepad باز نمایید و در اولین سطر عبارت /* Diploid SSR Data Set */ را دقیقا عین عبارت وارد نمایید( علامت های /* و */ بسیار حائز اهمیت هستند).
در سطر بعدی عبارت Number of populations = 3 را وارد نمایید چون تعداد جمعیت ما برابر سه است.
در سطر بعدی عبارت Number of loci = 3 را وارد نمایید چون ما سه لوکوس و یا مارکر را بررسی مینماییم.
در سطر بعد عبارت Locus name: را وارد نمایید
در سطر بعدی اسم لوکوس ها را وارد نمایید بین هر لوکوس یک Space فاصله بیندازید

شکل فوق: حال نوبت میرسد به وارد کردن جمعیت ها و ژنوتیپ افراد. در یک سطر اسم جمعیت اول را به صورت NAME = population1 وارد کنید و در سه سطر بعدی به ترتیب ژنوتیپ افراد مربوط به این جمعیت را وارد نمایید (سطر اول نفر اول، …..). و سپس همانند شکل فوق تمام جمعیت ها را وارد نمایید توجه داشته باشید که بین جمعیت های مختلف یک سطر فاصله باشد. سپس فایل را ذخیره نمایید. اکنون فایل ورودی داده ها آماده می باشد.
نحوه آماده سازی داده های مارکرهای Dominant مانند ISSR، و RAPD
در این مارکر ها ما فقط دو حالت داریم وجود آلل برای فرد (که آن را با عدد 1 نشان میدهیم) و یا عدم وجود الل برای فرد (که ان را با عدد 0 نشان میدهیم) به شکل زیر توجه نمایید
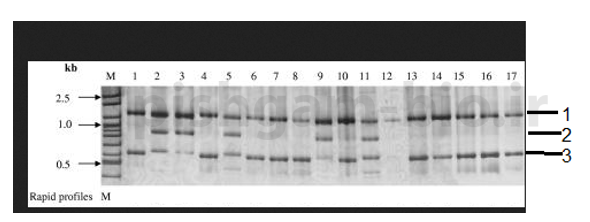
در این شکل 17 نفر با سه لوکوس RAPD مورد بررسی قرار گرفته اند. ژنوتیپ این افراد به این صورت می باشد:

عبارت 101 برای فرد شماره 1 به این معنی است که این فرد در لوکوس شماره
1 و 3 دارای الل بوده و در لوکوس شماره 2 فاقد الل بوده.
نحوه ساختن فایل ورودی برای این گونه مارکر ها نیز شبیه مارکر های co-domonant می باشد.
در شکل های زیر چند نمونه از فایل های آماده شده برای مارکرهای مختلف را مشاهده می فرمایید:

آموزش نحوه وارد کردن داده ها به نرم افزار popgene برای آنالیز فیلوژنتیکی
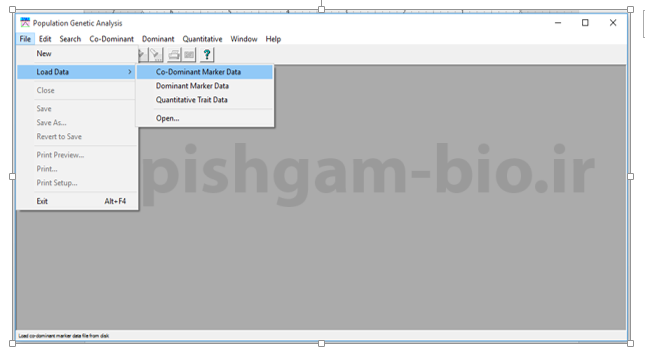
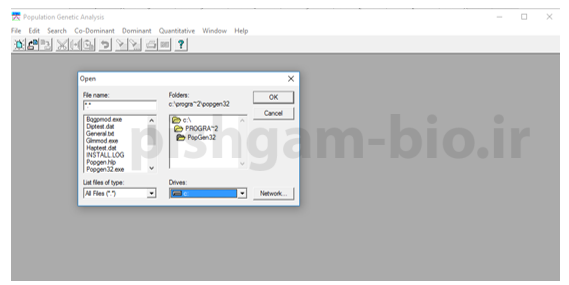
شکل فوق: برای وارد نمودن داده به نرم افزار از منوی File گزینه load data را انتخاب نموده و سپس انتخاب کنید داده ورودی از چه نوعی است.
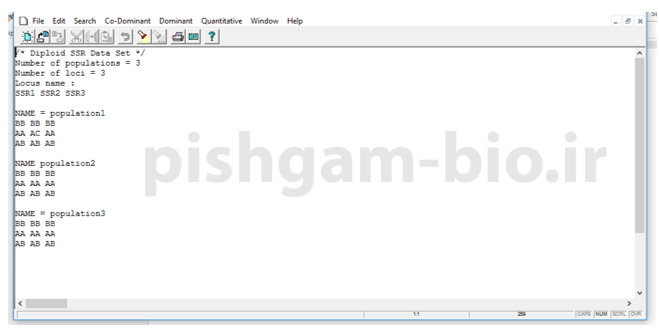
نحوه آنالیز داده ها در نرم افزار popegene
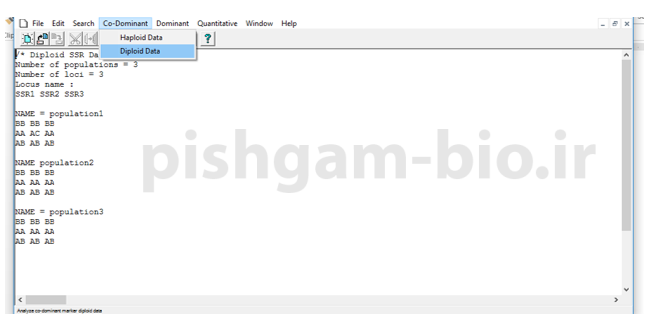
برای بررسی داده ها از منوها بالایی نرم افزار نوع داده خود dominant یا co-dominant را انتخاب نمایید سپس ژنوتیپ افراد مانند haploid و یا diploid را انتخاب نمایید
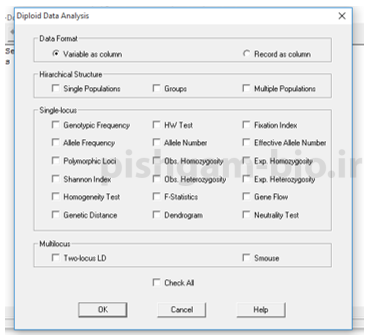
برای افراد دیپلوئید این صفحه به نمایش درخواهد امد که شما با انتخاب پارامترهای خود می تواند ان ها را در هر یک از جمعیت ها ( گزینه single population) و یا کل جمعیت محاسبه نمایید.
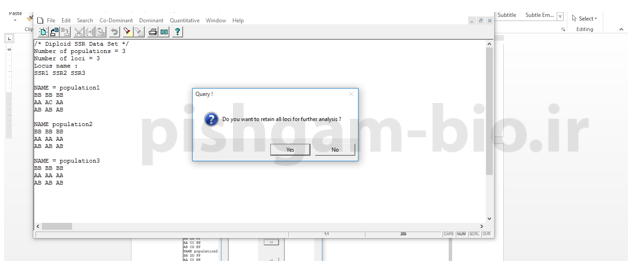
در صورتی که میخواهید همه لوکوس ها را بررسی نمایید گزینه yes را انتخاب نمایید اما در صورتی که میخواهید برخی لوکوس ها را از بررسی حذف نمایید گزینه no را بزنید تا به صفحه شکل زیر وارد شوید.
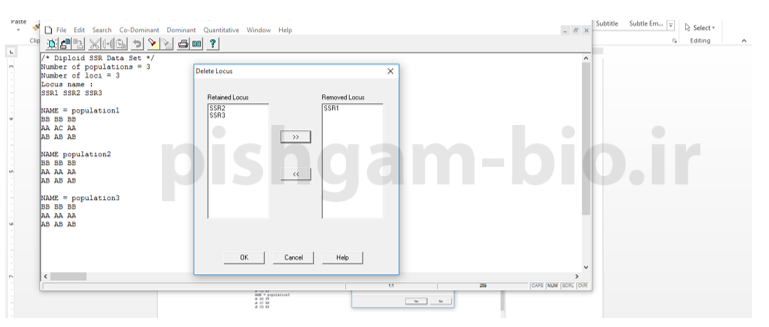
در این صفحه با انتقال لوکوس ها به کادر سمت راست تعیین میکنید که این لوکوس ها بررسی نگردند.
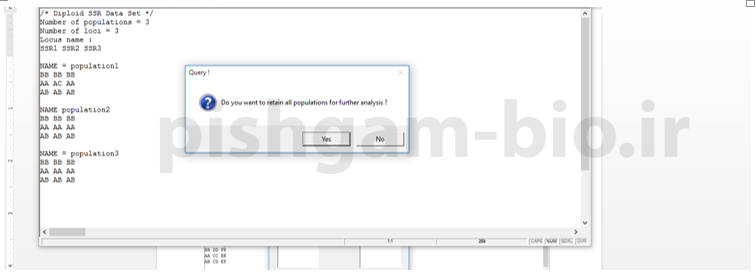
در صورتی که میخواهید همه جمعیت ها را بررسی نمایید گزینه yes را انتخاب نمایید اما در صورتی که میخواهید برخی جمعیت ها را از بررسی حذف نمایید گزینه no را بزنید تا به صفحه شکل زیر وارد شوید.
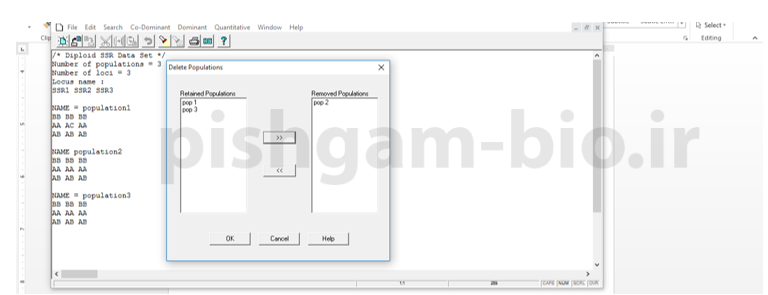
در این صفحه با انتقال جمعیت ها به کادر سمت راست تعیین میکنید که این جمعیت ها بررسی نگردند.
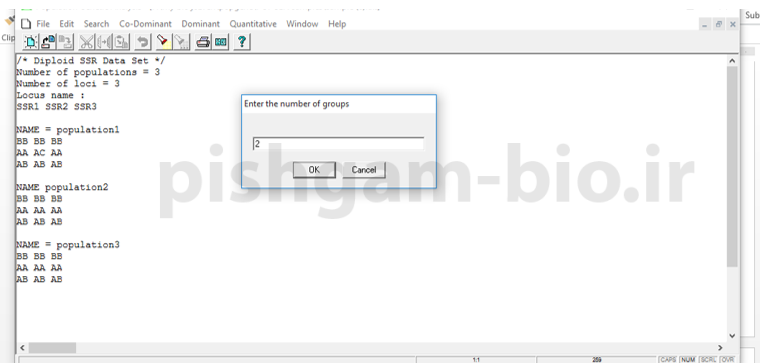
در صورتی که بخواهید جمعیت ها را در دو یا چند گروه تقسیم بندی کنید ( مثلا به خاطر جمع اوری نمونه از مناطق مختلف) در این کادر تعداد گروه ها را وارد نمایید. و در شکل زیر گروه بندی جمعیت ها را تعیین کنید.
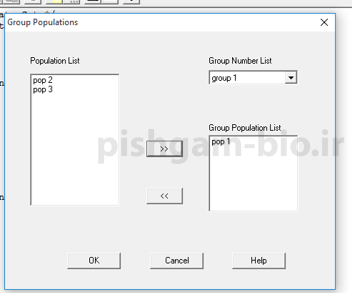
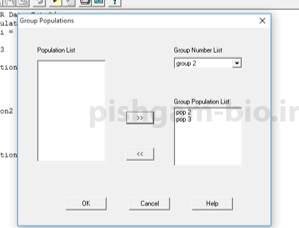
در نهایت برروی ok کلیک کنید تا آنالیزها انجام شوند و نتایج آن ها در صفحه بعدی نشان داده شود.
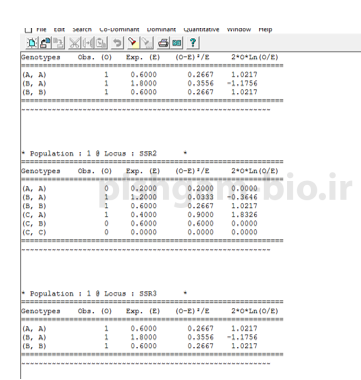
فراوانی ژنوتیپی هر لوکوس در جمعیت شماره یک
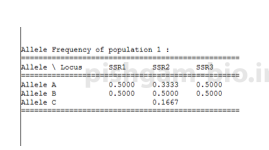
فراوانی اللی هر لوکوس در جمعیت شماره یک
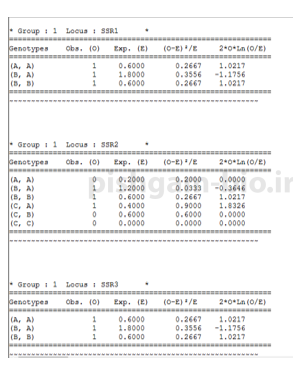
فراوانی زنوتیپی هر لوکوس در گروه شماره یک
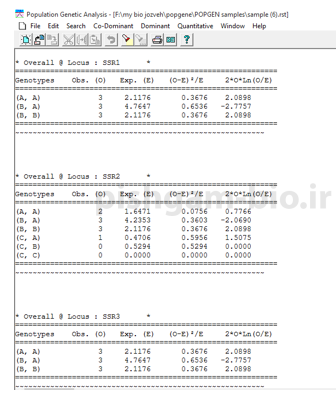
فراوانی ژنوتیپی هر لوکوس در کل جمعیت
آموزش محاسبه فراوانی ژنی و آللی، تعداد آلل ها، Effective Allele Number ، تنوع ژنی ، شاخص هموژنی ، شاخص شانون (Shannon)، ضریب فاصله ژنتیکی (Distance) ، تعادل هاردی واینبرگ در نرم افزار popgene
Gene Frequency: Estimates gene frequencies at each locus from raw data. Missingvalues are excluded from such estimation.
Allele Number: Counts the number of alleles with nonzero frequency.
Effective Allele Number: Estimates the reciprocal of homozygosity (Hartl and Clark1989, p.125).
Polymorphic Loci: Percentage of all loci that are polymorphic regardless of allelefrequencies.
Gene Diversity: Estimates Nei’s (1973) gene diversity.
Shannon Index: Estimates Shannon’s information index as a measure of gene diversity.
Homogeneity Test: Constructs two-way contingency tables and carries out chi-square(c2) and likelihood ratio (G2) tests for homogeneity of gene frequencies acrosspopulations. The tests are carried out for Groups or Multiple Populations.
F-Statistics: Estimates Nei’s (1973) GST for Groups or Multiple Populations, andestimates both GST and GCS for Groups and Multiple Populations.
Gene Flow: Estimates gene flow from the estimate of GST or FST (Slatkin and Barton1989). The estimation is made for Groups or Multiple Populations.
Genetic Distance: Estimates Nei’s (1972) genetic identity and genetic distance and Nei’s(1978) unbiased genetic identity and genetic distance. The estimation is made forGroups or Multiple Populations.
Dendrogram: Draws a dendrogram based on Nei’s genetic distances using UPGMA.This program is an adoption of program NEIGHBOR of PHYLIP version 3.5c by JoeFelsenstein. The drawing is executed for Groups or Multiple Populations.
Neutrality Test: Performs the Ewens-Watterson test for neutrality using the algorithmgiven in Manly (1985).
Two-locus LD: Estimates gametic disequilibria between pairs of loci and P2 tests forsignificance (Weir 1979) for Single Populations, and performs Ohta’s (1982a, b) twolocusanalysis of population subdivision (D-Statistics) for Multiple Populations.
Brown: Compute observed and expected moments of K, the number of heterozygous locibetween two randomly chosen gametes in a population, as well as multilocus indices and95% confidence limits from these moments (Brown et al., 1980) for Single Populations,and partition the total and average variances of K in a mixed pool of several populationsinto single-locus and two-locus components (Brown and Feldman 1981) for MultiplePopulations.
Smouse: Codes the most frequent allele as one (1) and a “synthetic” allele consisting ofall the other alleles combined as zero (0) (Yang and Yeh 1993). Estimates averageinterlocus correlation based on the coded data in a population (Smouse and Neel 1977)for Single Populations and estimates among- and within-population interlocuscorrelations for Multiple Populations.
اصلاعاتی که توسط این نرم افزار می توان از جمعیت ها به دست اورد: برای افراد دیپلوئید:
Genotypic Frequency: Estimates genotypic frequencies observed at each locus from rawdata only for co-dominant markers. Missing values are excluded from such estimation.
HW Test: Computes expected genotypic frequencies under random mating using thealgorithm by Levene (1949), and perform chi-square (c2) and likelihood ratio (G2) testsfor Hardy-Weinberg equilibrium at each locus only for co-dominant markers.
Fixation Index: Estimates FIS as a measure of heterozygote deficiency or excess (Wright1978) only for co-dominant markers.
Allele Frequency: Estimates gene frequencies at each locus from raw data. Missingvalues are excluded from such estimation.
Allele Number: Counts the number of alleles with nonzero frequency.
Effective Allele Number: Estimates the reciprocal of homozygosity (Hartl and Clark1989, p.125).
Polymorphic Loci: Percentage of all loci that are polymorphic regardless of allelefrequencies.Obs. Homozygosity: Estimates proportion of observed homozygotes at a given locus.
Exp. Homozygosity: Estimates proportion of expected homozygotes under randommating (see Exp. Heterozygosity for appropriate references).
Obs. Heterozygosity: Estimates proportion of observed heterozygotes at a given locusonly for co-dominant markers.
Exp. Heterozygosity: Estimates proportion of expected heterozygotes under randommating only for co-dominant markers. Two estimates are given. The first is Nei’s(1973) heterozygosity. The second the expected heterozygosity estimated using thealgorithm of Levene (1949), which is the same as Nei’s (1978) unbiased heterozygosity.
Shannon Index: Estimates Shannon’s (1949) information index as a measure of genediversity.
Homogeneity Test: Constructs two-way contingency tables and carries out chi-square(c2) and likelihood ratio (G2) tests for homogeneity of gene frequencies acrosspopulations. The tests are carried out for Groups or Multiple Populations.
F-Statistics: Estimates F-statistics (FIT, FST and FIS) for Groups or Multiple Populations(Hartl and Clark 1989), but estimates F-statistics for a three-level sampling hierarchy inrandom populations using a quite different approach by Weir (1990) for both Groups andMultiple Populations.
Gene Flow: Estimates gene flow from the estimate of GST or FST (Slatkin and Barton1989). The estimation is made for Groups or Multiple Populations.
Genetic Distance: Estimates Nei’s (1972) genetic identity and genetic distance and Nei’s(1978) unbiased genetic identity and genetic distance. The estimation is made forGroups or Multiple Populations.
Dendrogram: Draws a dendrogram based on Nei’s genetic distances using UPGMA.This program is an adoption of program NEIGHBOR of PHYLIP version 3.5c by JoeFelsenstein. The drawing is executed for Groups or Multiple Populations.
Neutrality Test: Performs the Ewens-Watterson test for neutrality using the algorithmgiven in Manly (1985).
Two-locus LD: Estimates Burrows’ composite measure of linkage disequilibria betweenpairs of loci and (c2) tests for significance (Weir 1979) for Single Populations, andperforms Ohta’s (1982a, b) two-locus analysis of population subdivision (D-Statistics) forMultiple Populations. Only for co-dominant markers.
Smouse: Codes a homozygote for the most frequent allele as one (1), a homozygote forthe “synthetic” allele consisting of all the other alleles combined as zero (0) and theirheterozygote as one-half (1/2). Estimates average interlocus correlation based on thecoded data in a population and test for both Hardy-Weinberg and linkage disequilibria(Smouse et al. 1983) for Single Populations and estimates among- and within-populationinterlocus correlations for Multiple Populations. In Multiple Populations case, noattempt was given to estimate Hardy-Weinberg disequilibrium in a subdivided populationas these estimates would be equivalent to F-statistics given above. Only for co-dominantmarkers.




دیدگاهتان را بنویسید